Parse Biosciences Evercode™ WT FFPE
/in Evercode, Parse, Partners, Seminar/by Harshita SharmaParse Biosciences Evercode™ WT FFPE
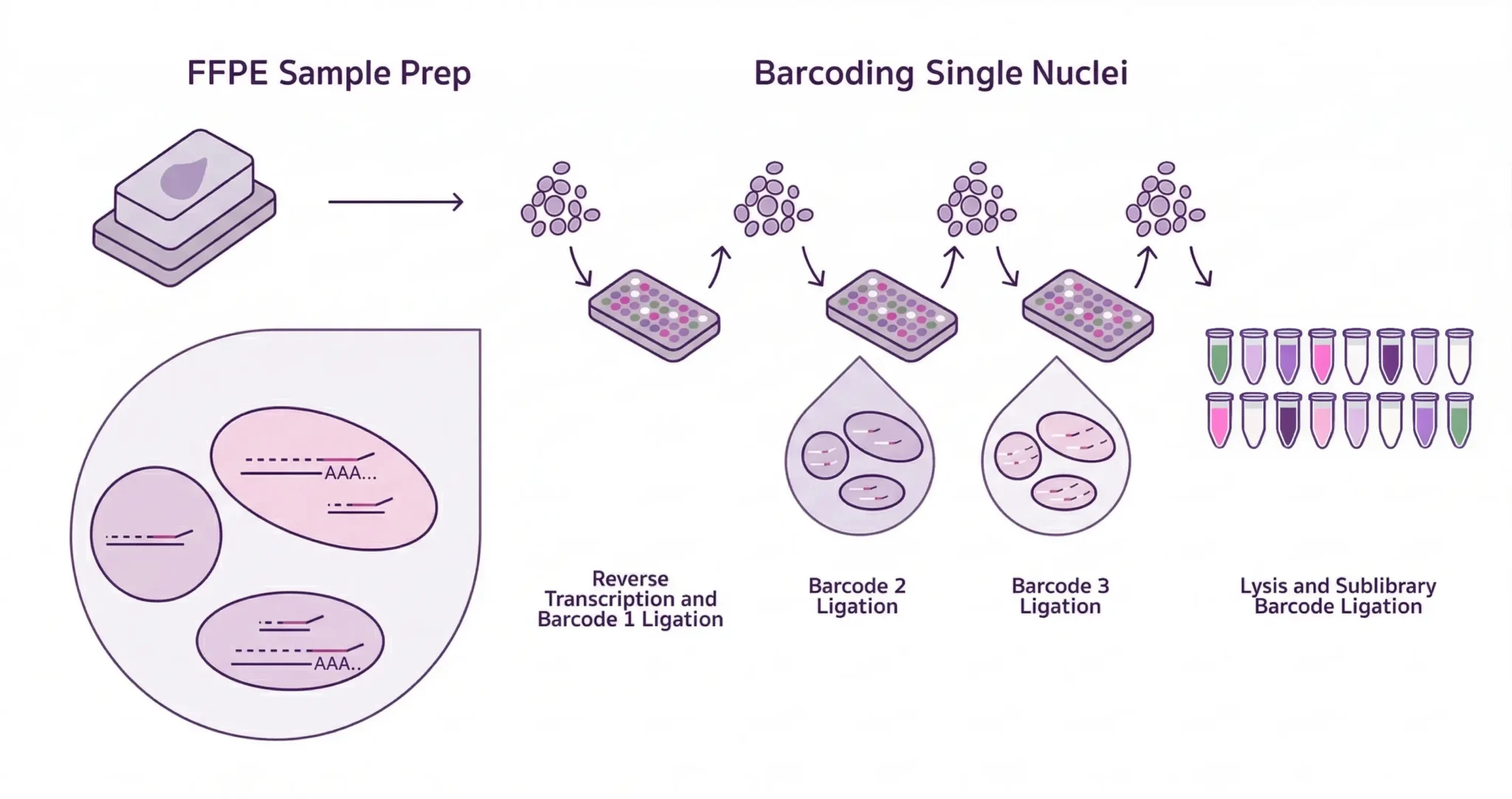
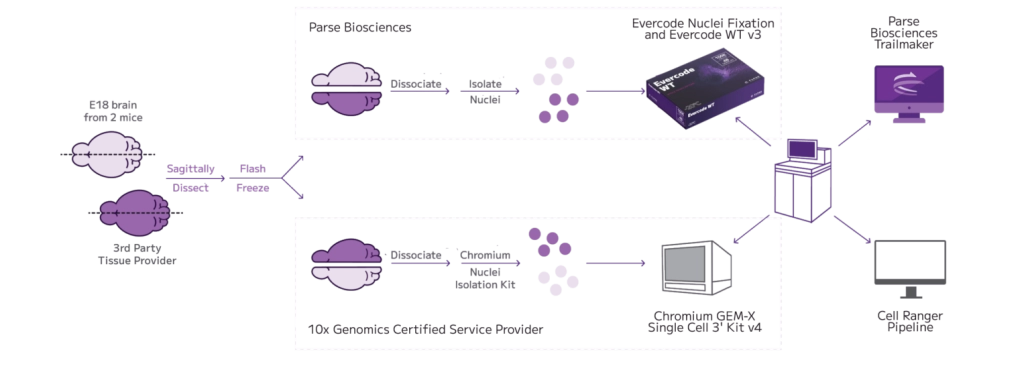
Evercode™ WT FFPE brings whole transcriptome single-cell RNA sequencing to formalin-fixed, paraffin-embedded tissue — unlocking retrospective cohorts that have historically been out of reach for discovery-driven transcriptomics. Unlike existing FFPE single-cell methods that rely on targeted probe panels and limit your analysis to a predefined gene list, Evercode WT FFPE uses reverse transcription-based chemistry to capture the full transcriptome directly from archived tissue. The result is unbiased, discovery-scale snRNA-seq from the clinical samples you already have in storage — without restricting what you can find before you start looking.
Designed for cohort-scale studies, Evercode WT FFPE supports processing from a single sample up to 96 samples per run, with cell throughput scaling from 10,000 to 5 million cells across four kit configurations. Combinatorial barcoding is built into the workflow — multiplexing requires no additional steps or protocols, minimising batch effects and technical variability across large sample sets. The platform is instrument-free and automation-compatible, supporting straightforward adoption and scale-up across standard and high-throughput laboratory environments. As your ANZ distributor for Parse Biosciences, Decode Science can advise on kit selection, experimental design, and workflow integration for your specific cohort.
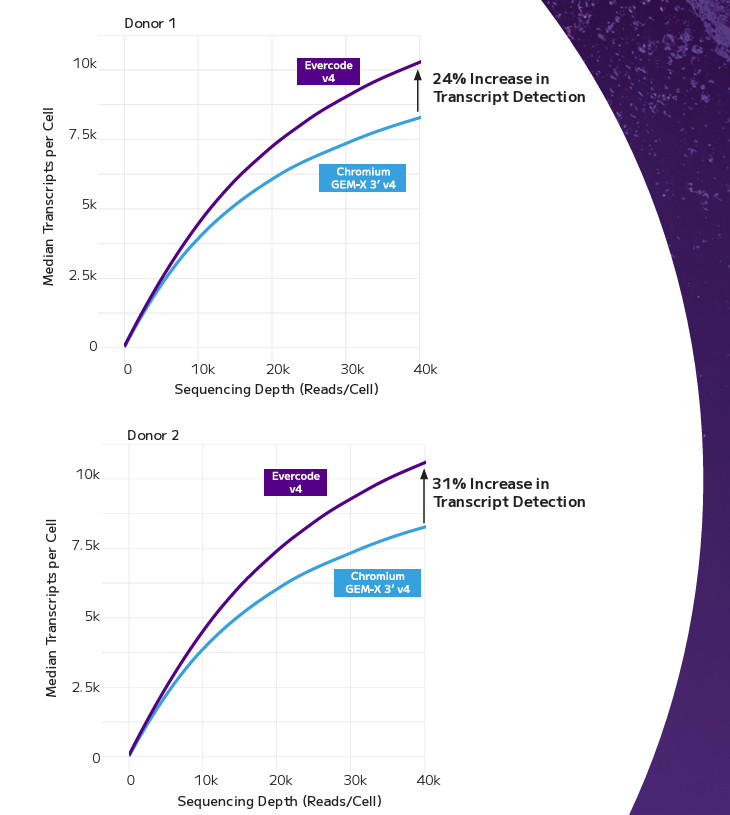
Whole Transcriptome Profiling — Not a Probe Panel
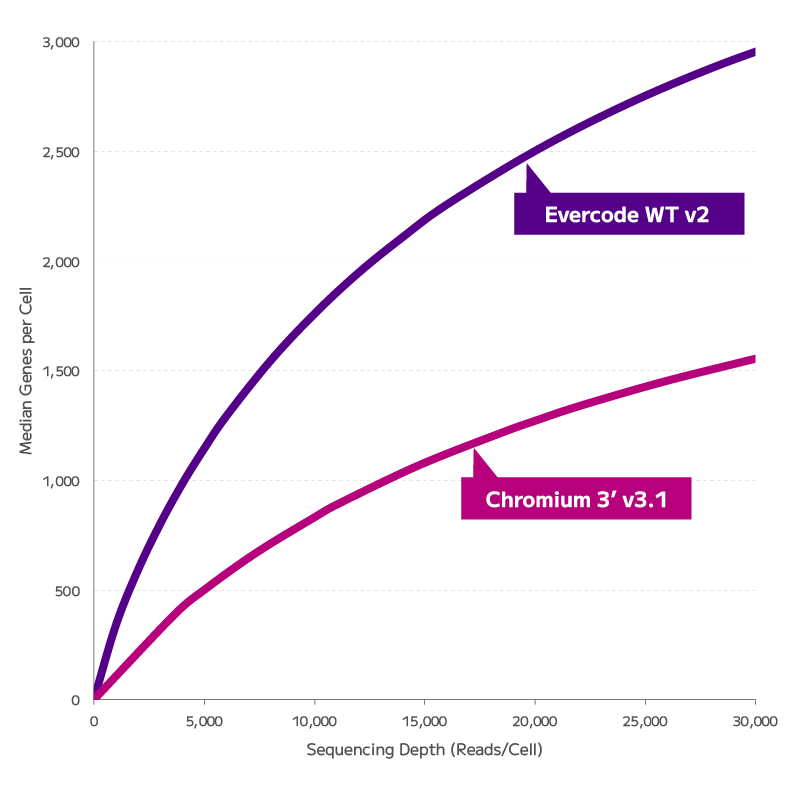
Every targeted FFPE single-cell method makes the same trade-off: you get clean data on the genes you chose, and nothing on the ones you didn't. Evercode WT FFPE captures the full transcriptomic landscape — including transcript isoforms, SNPs, and long non-coding RNAs — without restricting discovery to a predefined panel. For researchers working on cell state identification, regulatory biology, or exploratory cohort analysis, this is the difference between confirmation and genuine discovery.
Reliable Gene Expression Data From Degraded RNA
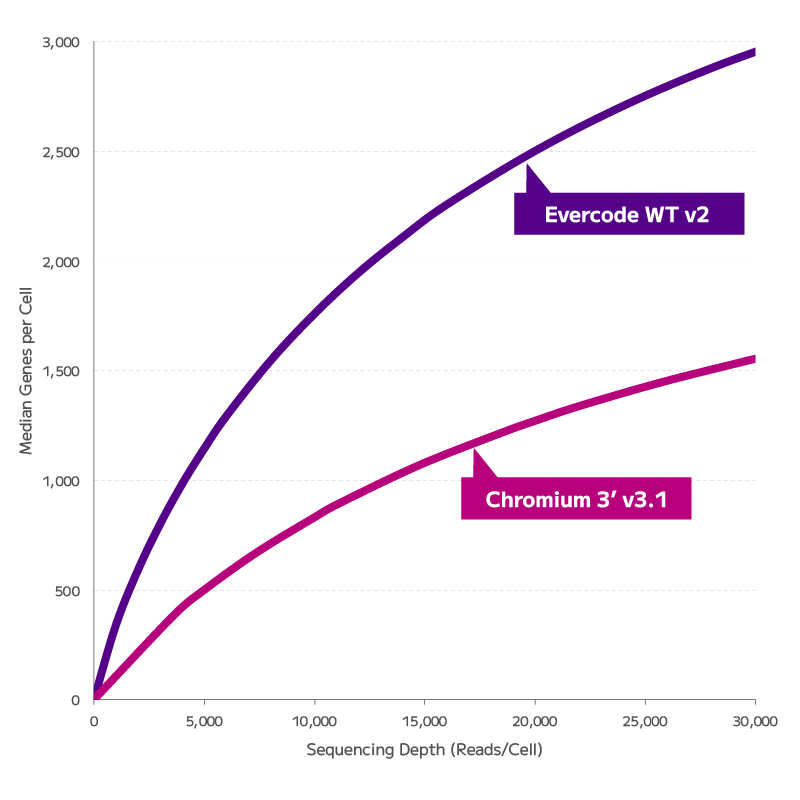
FFPE fixation fragments RNA, and fragmented RNA has historically degraded the accuracy of gene expression measurements. Evercode WT FFPE uses reverse transcription chemistry specifically optimised for fragmented RNA, enabling consistent gene expression detection and confident cell state identification from archival material. The resulting data are directly comparable with fresh tissue datasets — preserving biological signal across sample types and supporting cross-cohort integration without correction artefacts.
Cohort-Scale Multiplexing Built Into the Workflow
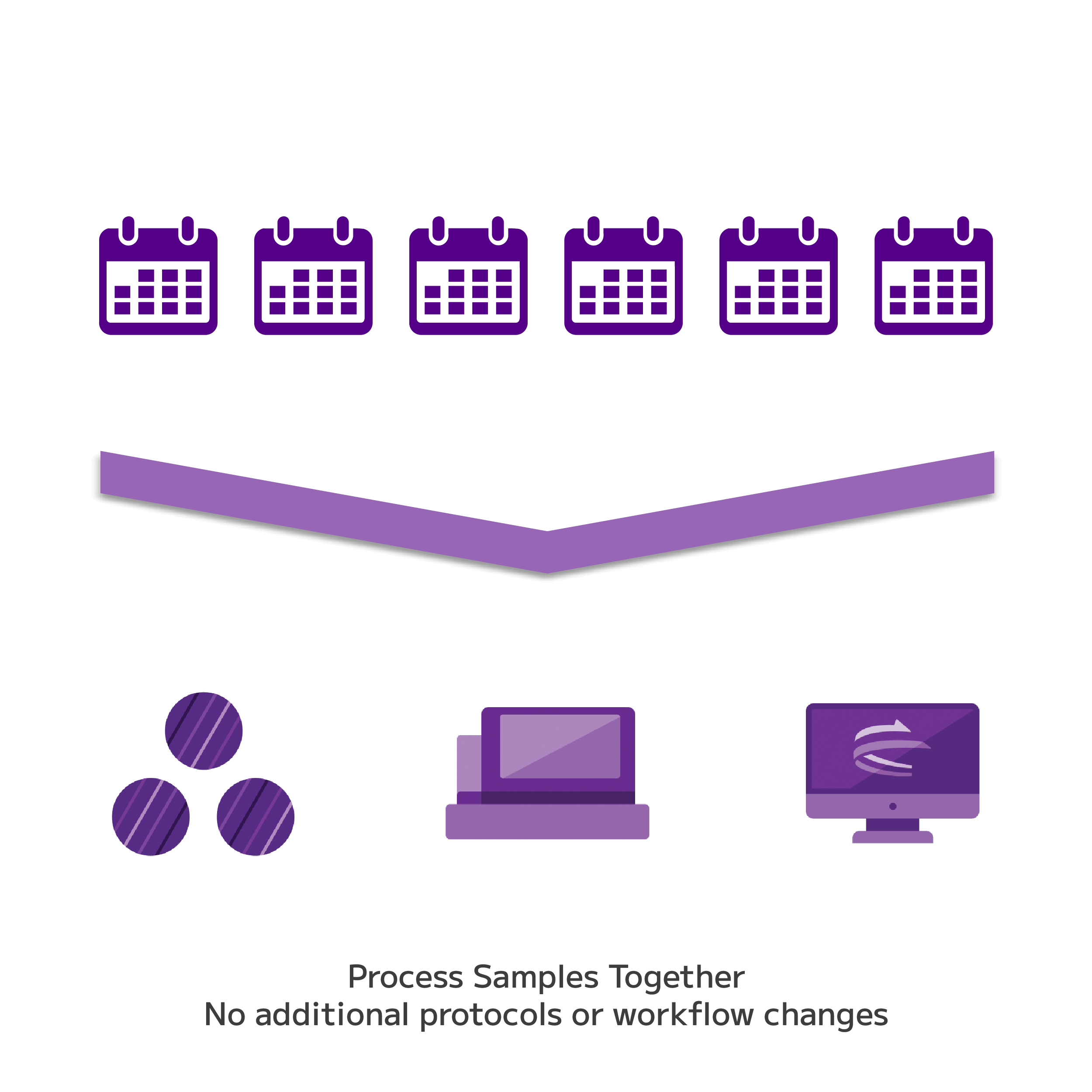
Combinatorial barcoding allows entire sample cohorts to be processed together in a single experiment — up to 96 samples per run — without additional multiplexing steps or reagents. This minimises technical variability between samples, supports robust statistical power across large study designs, and makes retrospective cohort studies at scale practically feasible for the first time with FFPE single-cell data.
Instrument-Free and Automation-Compatible Across Four Kit Sizes
Evercode WT FFPE requires no dedicated instrument for the core workflow, lowering the barrier to adoption and making it accessible to labs without specialised single-cell infrastructure. Four kit configurations — Mini, Standard, Mega, and Penta — scale from 10,000 cells and 1–12 samples up to 5 million cells and 1–96 samples, supporting everything from pilot experiments to large-scale clinical cohort studies. The workflow is compatible with laboratory automation for high-throughput operations.

Ebru Boslem, PhD
ANZ Market Manager - Research Genomics
Not sure which kit configuration fits your cohort size?
Our team can help you match the right Evercode WT FFPE kit to your sample numbers, cell input, and experimental design.
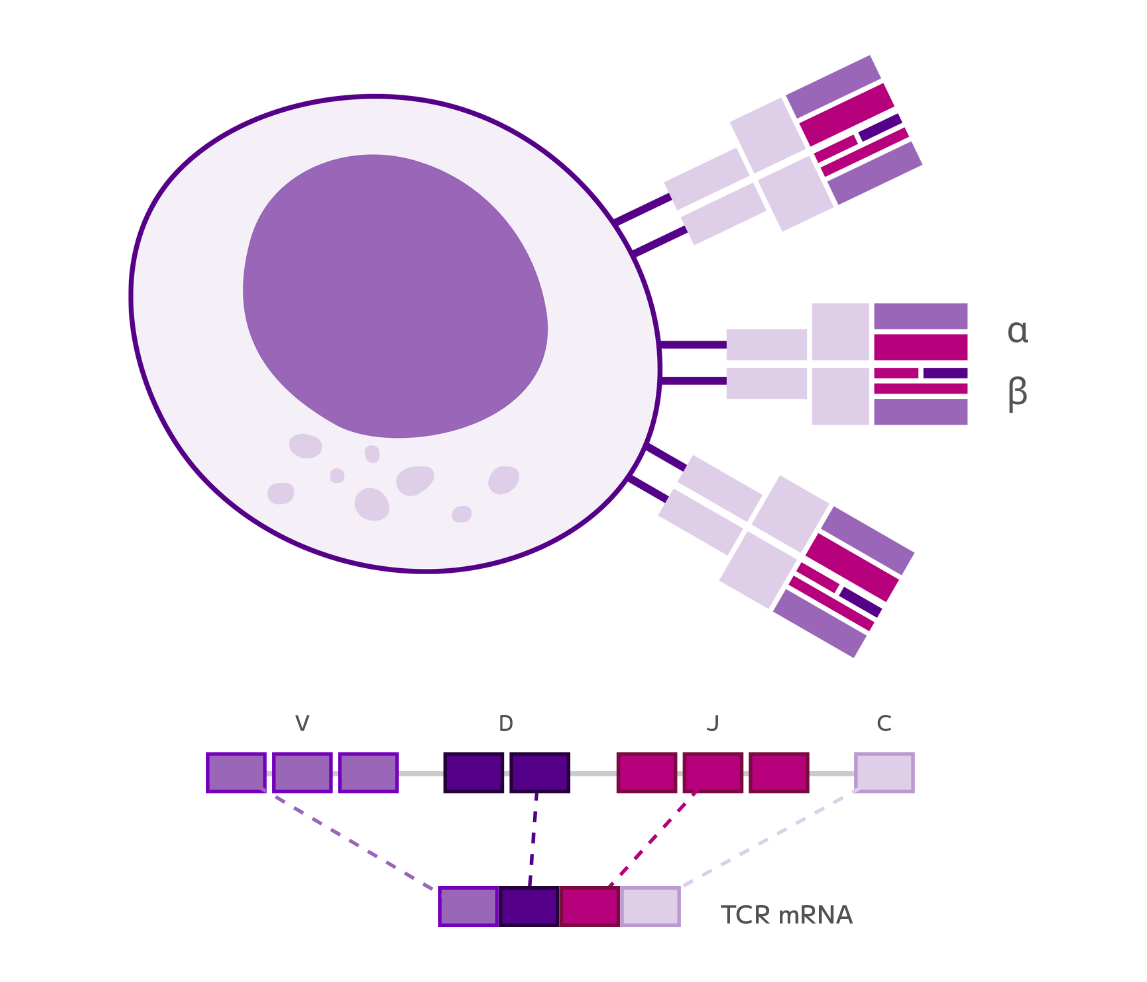
It all starts with Evercode split-pool combinatorial barcoding, our proprietary technology that labels molecules with cell-specific combinations of barcodes.
Because Your Most Valuable Samples Are Already in the Biobank
Fresh tissue single-cell studies are powerful — but they’re prospective by design. The patients you’ve already treated, the tumours you’ve already resected, the longitudinal samples you’ve already collected — those are locked in FFPE blocks, largely inaccessible to the transcriptomic methods that would extract the most value from them.
Evercode WT FFPE changes that calculus. Its relevance is clearest in these settings:
Retrospective cohort studies
Clinical biobanks represent years of carefully annotated patient samples. Whole transcriptome snRNA-seq from these collections enables discovery-driven analysis of disease progression, treatment response, and cellular heterogeneity at a scale and clinical depth that prospective studies take years to replicate.
Tumour microenvironment and cell state analysis
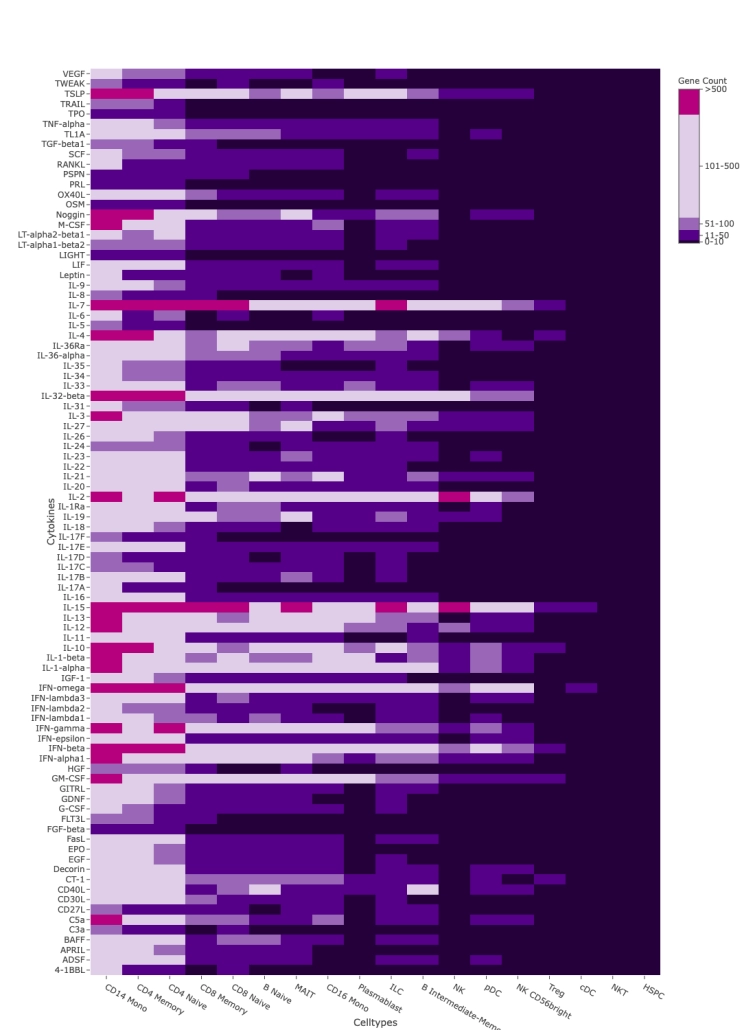
FFPE tissue preserves spatial and cellular context. Evercode WT FFPE recovers full transcriptomic complexity from these samples — including lncRNA expression and regulatory transcript diversity — enabling characterisation of rare cell states and microenvironmental programs that targeted panels miss entirely.
Multi-site and multi-cohort studies
Built-in multiplexing and instrument-free operation make it straightforward to harmonise sample processing across institutions and study sites, reducing the batch effects that complicate cross-cohort comparison.
Integration with fresh tissue data
Evercode WT FFPE data are directly comparable with fresh tissue snRNA-seq datasets, enabling combined analysis across sample types within a single study and supporting meta-analyses that span preservation methods.
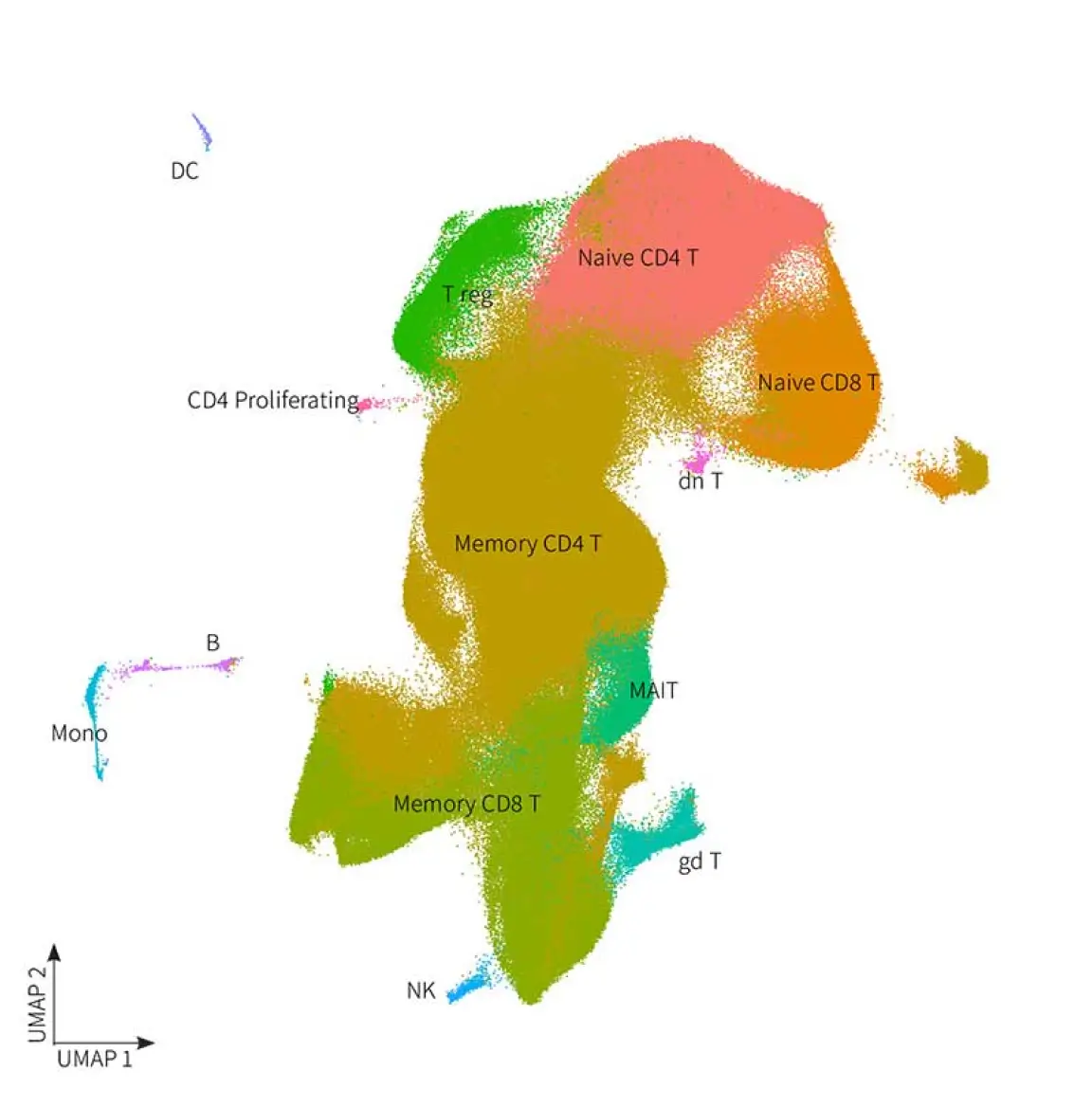
Accurate Cell State Identification From FFPE
Evercode WT FFPE enables confident identification and annotation of distinct cell populations from archived FFPE tissue, with gene expression measurements that support direct integration with fresh tissue reference datasets. Cell state resolution is maintained across sample types, preserving biological signal through the variability introduced by fixation and archival storage.
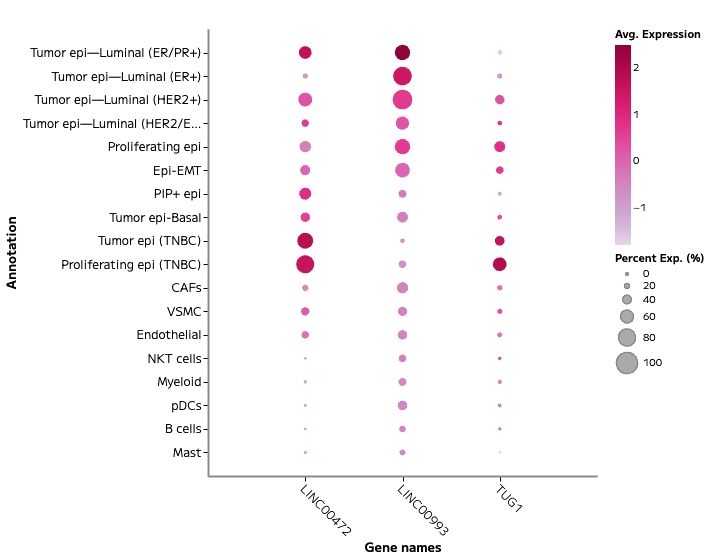
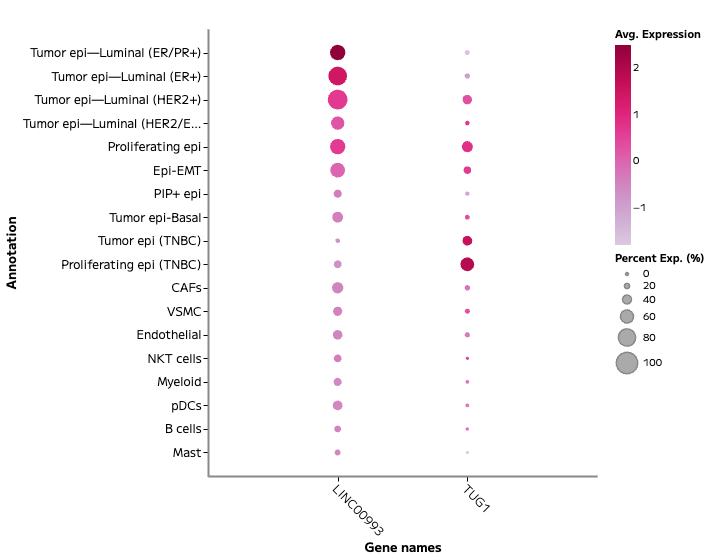
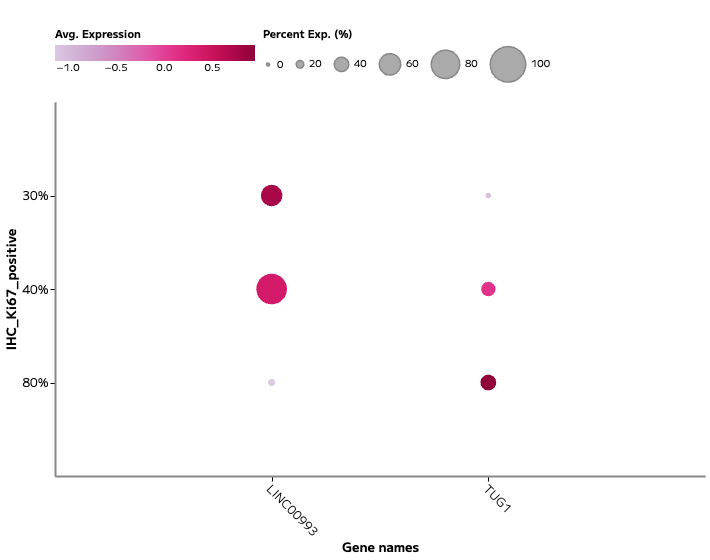
lncRNA and Regulatory Transcript Detection
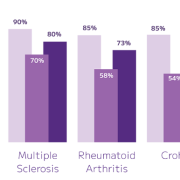
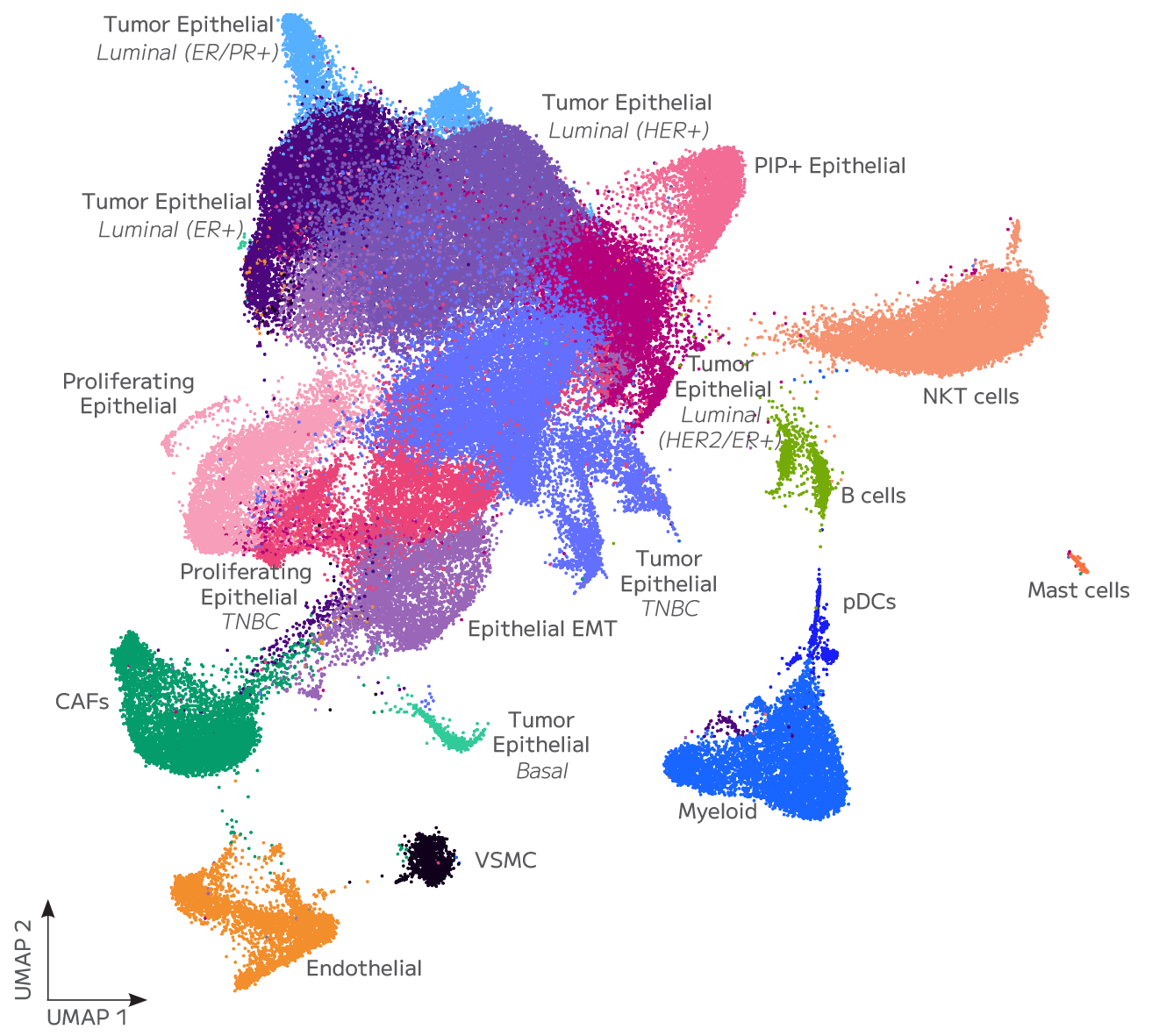
Whole transcriptome capture recovers biologically meaningful regulatory RNA species that targeted panels cannot access. Long non-coding RNA expression — including transcripts such as TUG1, LINC00993, and LINC00472 — is detectable across annotated cell populations, enabling analysis of regulatory programs within specific cell states. In TNBC tumour epithelial cells, for example, differential lncRNA expression across proliferating and non-proliferating states reflects the complex regulatory landscape across breast cancer subtypes — a finding that probe-based methods cannot surface.
Ready to sequence your FFPE cohort?
Request a quote or talk through your experimental design — we’ll respond at the earliest. Our team can help you match the right Evercode WT FFPE kit to your sample numbers, cell input, and experimental design.
Processing multiple samples within a single combinatorial barcoding experiment reduces technical variability and supports the statistical power needed for meaningful biological comparisons. Large retrospective cohorts can be processed together without introducing the batch effects that arise from sequential single-sample runs — a critical requirement for clinical cohort studies where sample-to-sample consistency directly impacts interpretation.
| Kit | Cells per Run | Samples per Run |
|---|---|---|
| Evercode WT FFPE Mini | Up to 10,000 | 1–12 |
| Evercode WT FFPE | 10,000–100,000 | 1–48 |
| Evercode WT FFPE Mega | 100,000–1,000,000 | 1–96 |
| Evercode WT FFPE Penta | 1,000,000–5,000,000 | 1–96 |
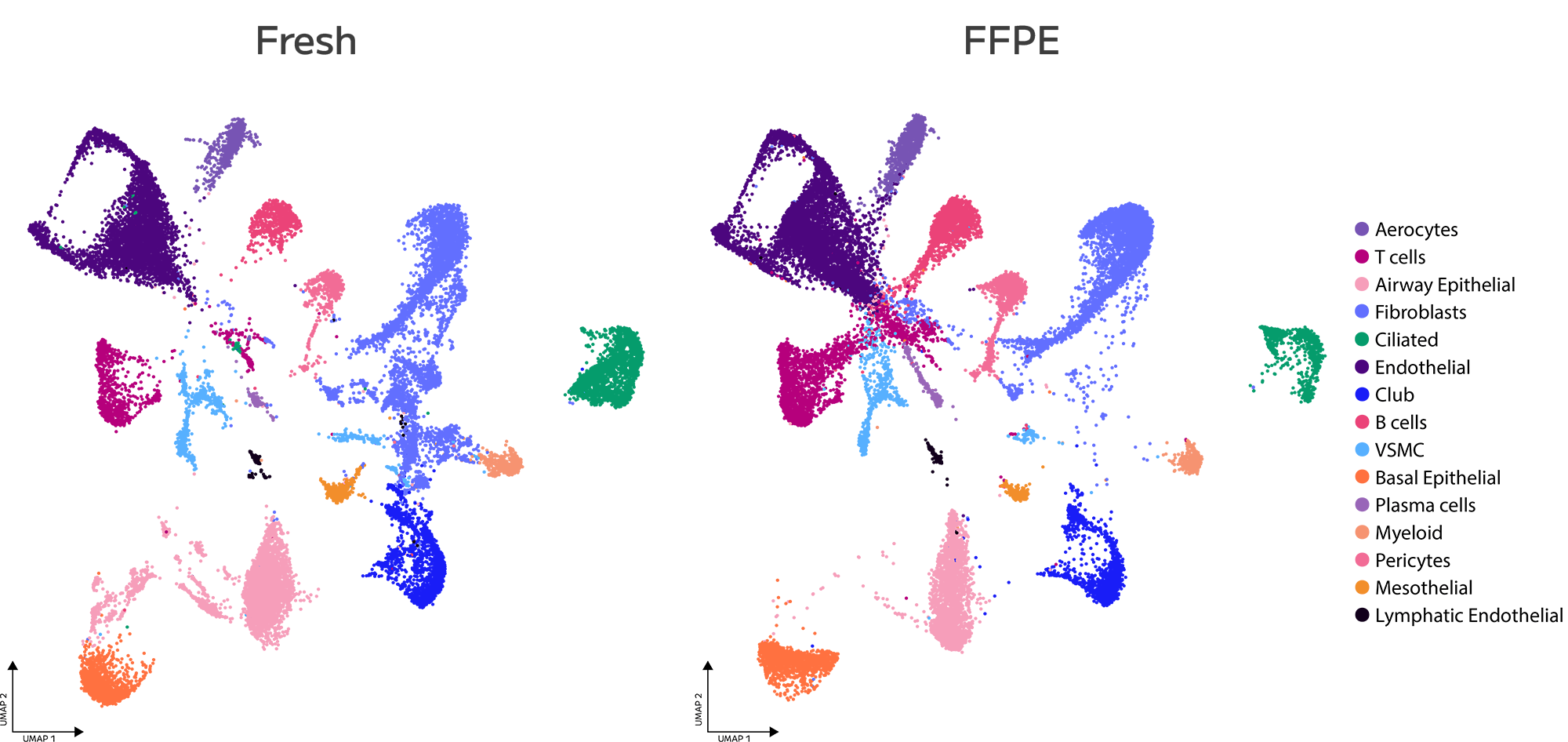
Download Product Sheet Instantly!

FAQs
What makes Evercode WT FFPE different from other FFPE single-cell methods?
Most existing FFPE single-cell approaches rely on targeted probe panels, which means your analysis is restricted to genes included in the panel at the time of design. Evercode WT FFPE uses reverse transcription-based chemistry to capture the full transcriptome, enabling unbiased discovery — including regulatory transcripts, lncRNAs, and transcript isoforms that probe-based methods cannot recover.
What tissue types has Evercode WT FFPE been validated on?
Evercode WT FFPE has been validated across a range of FFPE tissue types including tumour tissue. Contact Decode Science for tissue-specific guidance relevant to your sample type and storage conditions.
Do I need a dedicated instrument to run Evercode WT FFPE?
No. The core Evercode WT FFPE workflow is instrument-free and compatible with standard laboratory equipment. It is also compatible with laboratory automation for high-throughput processing.
How many samples can I process in a single run?
This depends on your kit configuration. The Mini kit supports 1–12 samples; the standard kit supports 1–48; and both the Mega and Penta kits support 1–96 samples per run.
Can Evercode WT FFPE data be integrated with fresh tissue snRNA-seq datasets?
Yes. Evercode WT FFPE is specifically designed to produce data directly comparable with fresh tissue datasets, supporting integration and cross-cohort analysis within a single study.
What sequencing platform is required?
Evercode WT FFPE libraries are compatible with Illumina sequencing platforms. Contact Decode Science for guidance on sequencing depth and read length requirements for your application.
What is the minimum RNA quality required?
Evercode WT FFPE uses reverse transcription chemistry optimised for fragmented RNA and does not require intact RNA. Contact Decode Science for guidance on tissue age, fixation duration, and input quality thresholds relevant to your samples.
Where can I access protocols and technical documentation?
Full protocols and technical documentation are available from Decode Science on request.
Our team is one form away.
We only need below information to serve you better. Decode Science respects your privacy and will never spam you with unrelated content.